Theoretical Study on the Regioselectivity of Electrophilic Aromatic Substitution Reactions of Azulene
Keywords:
Ab initio calculations, Azulene, Electrophilic Aromatic Reaction, NICSAbstract
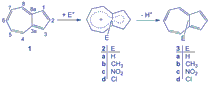
In this study electrophilic affinities were calculated at all reactive positions of azulene in electrophilic aromatic substitutions. Structures of cationic intermediates and products were optimized at HF/6-31+G* and B3LYP/6-31+G** levels of theory, and single point calculations were carried out at MP2/6-31+G**//B3LYP/6-31+G** for total energy. NICS calculations, activation energies and relative stabilities (deltaE) were used to predict the most reactive site in azulene. Results indicated that position 1 is the most reactive site for electrophilic aromatic reactions in terms of kinetic considerations, while the isomers with substituents at position 2 are predominantly thermodynamic products.
Downloads
Published
Issue
Section
License
Except where otherwise noted, articles in this journal are published under the Creative Commons Attribution 4.0 International License
