Computational thermochemical study of enthalpies of formation of B-alkylthiophenes using ab initio and DFT calculations
DOI:
https://doi.org/10.17344/acsi.2015.1683Keywords:
Computational thermochemical study, Standard molar enthalpies of formation, Atomization energy, Hess’s law, B-alkylthiophenesAbstract
The values for the standard molar enthalpies of formation of a series of the B-ring position alkyl-substituted thiophenes are calculated at 298.15 K using the Hartree-Fock (HF) and density functional theory (DFT) techniques. The results obtained are discussed in term of the substituent effect on the structural, electronic, and energetics of the titled molecules. The alkyl substitution with electron donating plays a fine-tune effect on the geometries, electronics, and energetics of the species. In the atomization energy route, the standard enthalpies of formation in the gas-phase, DHºf,298(g), obtained using the B3LYP/6-31G(d,p) method, can be successfully correlated to the substituent length via a linear dependence. However, DHºf,298(g), obtained using the HF/6-31G(d,p) method, is not able to predict their experimental behavior. In the formation reaction route, both the DFT and HF calculations reveal the same trend for predicting the values for the standard enthalpies of formation in the condensed-phase. It could be anticipated that the calculations can be extended to estimate the relative thermodynamic stabilities of oligo- and polymers consisting of this building blocks.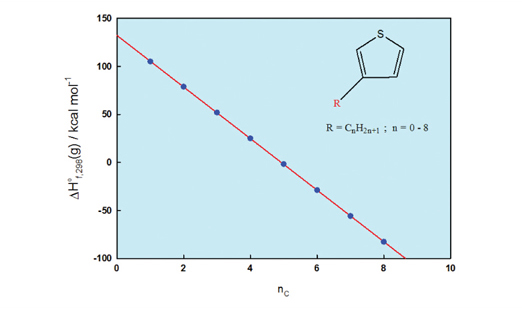
Downloads
Additional Files
Published
16.11.2015
Issue
Section
Physical chemistry
License
Except where otherwise noted, articles in this journal are published under the Creative Commons Attribution 4.0 International License
